Introduction:
Thalassemia disease is Understanding Thalassemia Beyond “Low Hemoglobin”
Many people ignore constant tiredness, pale skin, or shortness of breath, thinking, it is just weakness or iron deficiency. However, in some individuals, these signs point to a lifelong inherited blood disorder known as thalassemia disease.
Thalassemia disease is more than just “low blood.” It is a genetic condition that affects how the body produces hemoglobin, the substance that allows red blood cells to carry oxygen. This article explains thalassemia in a clear, student-friendly, and easy-to-understand way—covering its causes, types, symptoms, treatment options, and prevention strategies.
What Is Thalassemia?
Thalassemia is a genetic blood disorder in which the body produces abnormal or insufficient hemoglobin. Hemoglobin is a protein inside red blood cells responsible for transporting oxygen from the lungs to tissues and organs.
When hemoglobin production is faulty:
- Red blood cells become fragile.
- They break down faster than normal.
- Oxygen delivery to the body decreases.
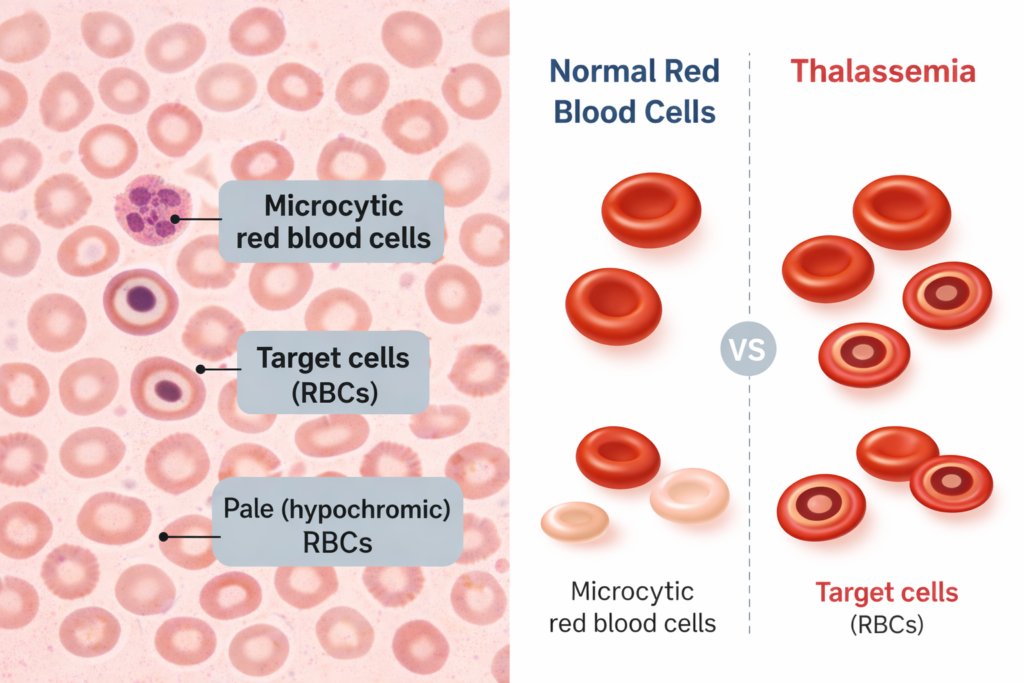
This leads to chronic anemia, which may range from mild to life threatening depending on the type of thalassemia.
Importantly, thalassemia is not an infectious disease. A person is born with it due to inherited genes from parents.
What Causes Thalassemia?
The cause of thalassemia lies in mutations of genes that control hemoglobin production. These genes are inherited from parents.
Genetic Inheritance Pattern:
- If one parent carries the thalassemia gene → the child may become a carrier (trait).
- If both parents are carriers → each pregnancy has:
- 25% chance of thalassemia major.
- 50% chance of being a carrier.
- 25% chance of being completely normal.
Thalassemia is not caused by:
- Poor nutrition.
- Iron deficiency.
- Infection.
- Lifestyle habits.
Types of Thalassemia
Thalassemia is classified based on which hemoglobin chain is affected.
1. Alpha Thalassemia
Alpha thalassemia occurs when the body cannot properly produce alpha globin chains. Humans normally have four alpha globin genes.
|
Type |
Gene Defect |
Clinical Features |
|
Silent Carrier |
1 gene missing |
No symptoms |
|
Alpha Thalassemia Trait |
2 genes missing |
Mild anemia |
|
Hemoglobin H Disease |
3 genes missing |
Moderate to severe anemia |
|
Alpha Thalassemia Major |
4 genes missing |
Usually fatal without early intervention |
Beta thalassemia affects the production of beta globin chains and is more commonly diagnosed.
Type | Severity | Characteristics |
Beta Thalassemia Minor (Trait) | Mild | Often asymptomatic |
Beta Thalassemia Intermedia | Moderate | Occasional transfusions |
Beta Thalassemia Major | Severe | Regular transfusions required |
Beta thalassemia major, also called Cooley’s anemia, usually appears within the first two years of life.
Signs and Symptoms of Thalassemia disease
Symptoms depend on the type and severity of the disease.
Common Symptoms
- Chronic fatigue and weakness.
- Pale or yellowish skin.
- Shortness of breath.
- Dizziness or headaches.
- Cold hands and feet.
Moderate to Severe Symptoms
- Delayed growth and puberty in children.
- Bone deformities (especially facial bones).
- Enlarged spleen and liver.
- Dark-colored urine.
- Frequent infections.
In Infants with Severe Thalassemia
- Poor feeding.
- Failure to thrive.
- Severe anemia within months of birth.
How Is Thalassemia Diagnosed?
Diagnosis is made through:
- Complete Blood Count (CBC).
- Peripheral blood smear.
- Hemoglobin electrophoresis.
- Genetic testing (if required) .
Carrier screening is especially important in regions where thalassemia disease is common, including South Asia and Nepal.
Treatment of Thalassemia
Treatment of thalassemia is depends on disease severity and aims to maintain normal hemoglobin levels and prevent complications.
1. Mild Thalassemia (Trait)
- Usually no treatment required.
- Avoid unnecessary iron supplements.
- Regular monitoring advised.
2. Moderate to Severe Thalassemia
Blood Transfusions
- Required every 2–4 weeks.
- Maintain adequate hemoglobin levels.
Iron Chelation Therapy
- Removes excess iron from the body.
- It Prevents damage to heart.
- It liver, and endocrine organs.
Folic Acid Supplementation
- Supports red blood cell production.
Splenectomy (Selected Cases)
- Performed if spleen becomes overactive.
Bone Marrow / Stem Cell Transplant
- Only curative option available.
- Best results with matched sibling donor.
Gene Therapy (Emerging)
- Uses modified patient stem cells.
- Shows promising long-term results.
Prevention of Thalassemia
Because thalassemia disease is inherited, prevention focuses on awareness and screening.
Key Preventive Measures
- Premarital carrier screening.
- Genetic counseling.
- Prenatal diagnosis (CVS or amniocentesis).
- Community awareness programs.
Early detection helps families make informed reproductive decisions.
Living With Thalassemia
With modern medical care, individuals with thalassemia can:
- Attend school.
- Build professional careers.
- Start families.
- Live long and meaningful lives.
Regular follow-ups, emotional support, and proper treatment significantly improve quality of life.
Frequently Asked Questions (FAQs)
What is thalassemia trait?
Thalassemia trait (minor) means a person carries one defective gene. They usually have mild anemia or no symptoms but can pass the gene to children.
Why does thalassemia cause anemia?
Thalassemia causes abnormal hemoglobin production, making red blood cells fragile and short-lived, which leads to chronic anemia.
What is the difference between alpha and beta thalassemia?
- Alpha thalassemia affects alpha globin chains.
- Beta thalassemia affects beta globin chains.
The severity depends on how many genes are affected.
Can thalassemia patients get married and have children?
Yes. With proper medical guidance, thalassemia patients can marry and plan families safely.
How often do thalassemia major patients need blood transfusions?
Most patients require transfusions every 2–4 weeks to maintain safe hemoglobin levels.
Why is iron chelation therapy necessary?
- Repeated blood transfusions cause iron overload, which can damage the heart, liver, and glands. Chelation therapy removes excess iron from the body.
Can thalassemia patients live a normal life?
Yes. With proper treatment and regular follow-up, many patients live long, productive, and independent lives.
Can medicines alone cure thalassemia?
No. Medicines help manage symptoms and complications, but they do not correct the genetic defect.
Is thalassemia contagious?
- No. Thalassemia cannot spread through contact, food, blood, or air.